Les étapes qui mènent à
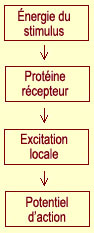
l'activation d'un nocicepteur sont les mêmes que pour les récepteurs
tactiles spécialisés. L'énergie du stimulus douloureux, qu'elle
soit mécanique, thermique ou chimique, modifie la forme de certaines
protéines situées dans la membrane cellulaire des fibres
A delta ou C. Ce changement de forme modifie la perméabilité
de la membrane dans le sens d'une excitation locale proportionnelle à l'énergie
du stimulus. Lorsque cette excitation atteint un
certain seuil, un influx nerveux ou potentiel
d'action est envoyé.
Et comme l'amplitude d'un potentiel
d'action est toujours la même, les variations d'intensité du stimulus
nociceptif se traduiront ensuite par la fréquence du train d'impulsions
nerveuses, le moyen de prédilection qu'utilisent les neurones pour communiquer
entre eux.
À chacune des synapses
le long de cette voie de la douleur, plusieurs neurotransmetteurs permettent la
transmission du message nociceptif. Les substances identifiées à
ce jour se répartissent en deux grands groupes principaux : les neurotransmetteurs
dits " classiques " et les neuropeptides.
Le premier groupe comprend entre autres le glutamate, l'aspartate et la
sérotonine. Au moins une vingtaine de peptides contribuent pour leur part
à transmettre l'influx douloureux : substance P, peptide intestinal vaso-actif,
peptide relié au gène de la calcitonine, somatostatine, cholécystokinine,
ACTH, etc. Sans compter la grande famille des enképhalines
dont les peptides exercent un effet inhibiteur dans les
voies de contrôles descendantes.
À noter qu'une même
fibre nociceptive peut contenir différents peptides et neurotransmetteurs
classiques. Leurs rôles respectifs restent d'ailleurs encore très
largement à déterminer. Il est également difficile de corréler
les propriétés électrophysiologiques des afférences
nociceptives à leur contenu en peptides.
Néanmoins,
le glutamate et la substance P, un peptide de 11 acides aminés
de la famille des tachykinines, semblent être parmi les substances les plus
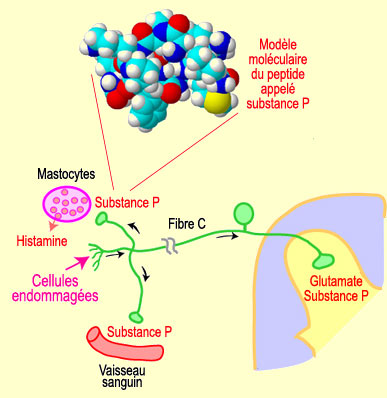
impliquées dans la transmission de la douleur. La substance P se fixe par
exemple sur des récepteurs
spécifiques appelés NK1 situés sur les neurones nocicepteurs
de la corne dorsale de la moelle épinière.
Elle
est également présente dans le cerveau où elle est associée
à la régulation des troubles de l'humeur, de l'anxiété,
des renforcements,
de la neurogenèse,
de la neurotoxicité, du rythme respiratoire, des nausées et, évidemment,
de la douleur. De plus, par l'intermédiaire d'un phénomène
appelé réflexe d'axone, la substance P peut aussi être libérée
au niveau périphérique sur le site d'une lésion tissulaire.
Elle provoque alors une forte vasodilatation qui peut induire la
libération de différentes substances (bradykinine, histamine,
sérotonine, etc).
En général, la substance P a été
associée à des connexions excitatrices plutôt lentes, et donc
à des douleurs chroniques persistantes véhiculées par les
fibres
C, tandis que le glutamate participe à des neurotransmissions rapides
caractéristiques de la douleur aiguë associée aux fibres A
delta. Leurs récepteurs peuvent être distribués dans différentes
populations de neurones préservant ces spécificités. Mais
ils peuvent aussi coexister sur les mêmes neurones comme on l'a observé
dans plusieurs régions du système nerveux central. De nombreuses
compagnies pharmaceutiques ont tenté de développer des antagonistes
de la substance P, espérant ainsi trouver un puissant analgésique.
Mais les résultats ont été très décevants.
Au niveau périphérique, d'autres
substances vont aussi contribuer à transmettre la douleur et à
rendre nos nocicepteurs plus sensibles. Elles proviennent de la lésion
tissulaire elle-même (par exemple les ions H+ et K+). Elles peuvent aussi
être liées au processus inflammatoire (leucotriènes, prostaglandines...)
et agir en sensibilisant les nocicepteurs aux agents précédents.
Ou encore elles sont libérées par les nocicepteurs eux-mêmes
et peuvent activer ces derniers directement ou indirectement (par exemple la substance
P).
Car parallèlement au contrôle
descendant de la douleur et à ses endorphines
qui nous permettent de tolérer un effort douloureux pour le corps ou qui
permet de se concentrer sur autre chose que sa douleur, les processus de sensibilisation
et d'inflammation tendent, eux, à nous faire immobiliser la partie du corps
blessée pour favoriser l'action de la " soupe inflammatoire "
de molécules et la guérison. Et " l'argument ", si l'on
peut dire, pour nous convaincre de faire attention à la région lésée
de notre corps, ce sont les sensations douloureuses que vont maintenant produire
de simples stimuli tactiles dans les environs de la blessure.
La sensibilisation
centrale dans la moelle épinière, qui peut amplifier encore davantage
la réponse douloureuse à un stimulus normal, utilise pour sa part
des mécanismes
cellulaires distincts.
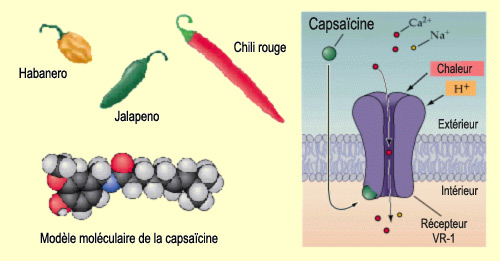
Des piments forts comme le jalapeno sont consommés
chaque jour par près du tiers de la population mondiale. Leur goût
piquant leur vient principalement de la capsaïcine, une molécule
qui provoque une sensation de brûlure en se fixant sur un
récepteur particulier appelé TRPV1, situé sur
nos nocicepteurs. Le récepteur TRPV1 peut également être activé
par la chaleur ou un composé endogène, l'anandamide,
qui active aussi nos récepteurs
aux cannabinoïdes. Chaleur et anandamide sont donc probablement
les activateurs naturels de ces récepteurs qui, par hasard, peuvent aussi
être activés par une molécule exogène d'origine végétale
comme la capsaïcine.
D'après
: Neuroscience, Dale Purves, Jean-Marie Coquery.
Les récepteurs
de la famille des vanilloïdes, comme les TRPV1, sont des récepteurs
canaux qui, lorsque stimulés, laissent entrer dans la cellule
du calcium et du sodium. D'où une dépolarisation du neurone qui,
si elle est suffisante, déclenche des potentiels
d'action. Le nocicepteur libère alors de la substance P qui
excite le neurone suivant de la voie ascendante nociceptrice et ainsi de suite
jusqu'au cerveau. C'est alors qu'on se rendra compte à quel point notre
repas est épicé !
Mais pourtant, quelques instants plus tard,
il arrive souvent qu'on le trouve finalement moins piquant qu'au tout début.
C'est que le contact prolongé de la capsaïcine avec son récepteur
désensibilise celui-ci. Ironiquement, la capsaïcine peut donc aussi
produire une analgésie causée en partie par une déplétion
en substance P. La capsaïcine est ainsi l'un des ingrédients principaux
de certaines crèmes analgésiques et anti-inflammatoires qui sont
utilisées contre de simples douleurs aux muscles ou aux articulations,
mais aussi contre des douleurs plus difficiles à traiter comme l'arthrite
et les douleurs
neuropathiques. Ces crèmes contiennent souvent un autre ingrédient
comme la lidocaïne qui permet de diminuer la sensation de brûlure initiale
due à la capsaïcine.
Les récepteurs de la capsaïcine
se retrouvent chez tous les mammifères, mais pas chez les oiseaux. Cela
a permis la mise au point de graines pour les mangeoires d'oiseaux à l'épreuve
des écureuils ! De plus, des souris dont le gène du récepteur
de la capsaïcine a été désactivé peuvent boire
une solution de capsaïcine comme si c'était de l'eau…
Les études phylogénétiques
ont montré l'importance des endorphines qui existent chez tous les vertébrés.
Certains pensent même qu'elles auraient pu servir à s'émanciper
des réactions de protection automatiques déclenchées par
un stimulus nociceptif. Et ce faisant, à favoriser des comportements qui,
dans plusieurs situations, s'avèrent plus adaptatifs.
Par exemple,
si une proie blessée s'arrête pour lécher sa blessure au lieu
de continuer à fuir malgré la douleur, ses chances de survie sont
réduites. Mais la sécrétion d'endorphines déclenchée
par la peur, le stress et la course va rendre cette douleur tolérable et
inciter l'animal à continuer sa course.
Les endorphines permettent
donc que la survie passe en premier et que la récupération et la
guérison viennent ensuite.
Depuis les
expériences de Jonathan Levine en 1978, on sait que des suggestions
psychologiques peuvent en effet déclencher la sécrétion d'endorphines
qui vont diminuer la perception douloureuse.
Les études subséquentes
ayant par la suite complexifié le tableau, il semble maintenant que l'effet
placebo a une composante endorphiniques et une composante non endorphiniques,
la première étant davantage liée aux attentes et l'autre
aux conditionnements.
Une fois que les endorphines se sont fixées
à leur récepteur et ont produit leur effet, elles sont rapidement
inactivées. Le principal mécanisme de cette inactivation est la
dégradation enzymatique par une famille d'enzymes appelées peptidases.
Et c'est en coupant les liens peptidiques qui relient les différents acides
aminés que les peptidases agissent.
En 2003, des chercheurs ont
isolé une substance, la sialorphine, qui est sécrétée
chez le rat et qui bloque l'action des enzymes de dégradation en s'y fixant.
Les enképhalines peuvent alors rester actives plus longtemps et il en résulte
un puissant effet antidouleur. Les rats auxquels on a injecté de la sialorphine
pouvaient ainsi se promener librement sur des surfaces cloutées.
Dans
des conditions naturelles, la sialorphine est libérée dans le sang
suite à un stress. Par exemple, chez les rats mâles soumis à
des conditions de
compétition et d'agression vis-à-vis de leurs congénères,
elle atténue la douleur des blessures.
Ces propriétés
intéressantes de la sialorphine de rat ont amené les chercheurs
à traquer son homologue fonctionnel chez l'être humain. Et quelques
années plus tard, une molécule équivalente, l'opiorphine,
fut découverte. Reste maintenant à savoir dans quelles situations
l'opiorphine est sécrétée chez l'humain et comment elle contribue
à l'effet analgésique des endorphines.
Par ailleurs, une
meilleure connaissance de tels bloqueurs naturels des peptidases pourrait aider
à la conception de nouveaux médicaments qui, en empêchant
la destruction des opioïdes endogènes, pourraient atténuer
la douleur.
L'opium
était sans doute déjà connu des Sumériens, environ
3 000 ans av. J.-C. comme en témoignent des tablettes gravées de
cette époque. Certains écrits égyptiens datant de l'époque
de Ramsès II (1300 av. J.C.) vantent expressément les vertus "dormitives"
et analgésiques de cette plante.
Français
fumeurs d'opium, couverture du Petit Journal du 5 juillet 1903
Mais
c'est à partir du XVIIIe siècle que celle-ci acquiert véritablement
ses lettres de noblesse. Un premier dérivé actif de l'opium est
décrit comme un alcaloïde végétal par F. W. Serturner
dans ses travaux publiés en 1805-1806 et 1817. Connaissant la somnolence
qu'il produisait sur l'être humain, Serturner le nomme morphium (en français,
morphine), en référence au dieu des rêves
de la Grèce antique, Morphée.
Ce n'est qu'en
1925 que la structure moléculaire complexe de la morphine est décrite
par le chimiste britannique Robert Robinson. Et à partir de 1952, il est
possible de synthétiser chimiquement la morphine ou ses dérivés.
Cette synthèse chimique donnera naissance à des composés
dont la structure est proche de la morphine, comme le dextrométhorphane
(analgésique analogue de la codéine), mais dont les effets diffèrent
quelque peu.
L'usage des substances opiacées se répand alors
très rapidement en médecine. Henri
Laborit élabore par exemple un cocktail injectable associant opiacés
et neuroleptiques. Utilisé durant la guerre d'Indochine, il facilite le
transfert des blessés vers les blocs opératoires.
Dans les
années 1970, les
découvertes successives de récepteurs spécifiques aux opioïdes
puis des enképhalines (les premières " morphines endogènes
") ouvre de nouvelles perspectives à notre compréhension de
la douleur et de ses mécanismes
de contrôle. Et non seulement par des molécules venant de l'extérieur
du corps (les médicaments), mais aussi par des molécules endogènes,
c'est-à-dire produites par nos propres neurones. L'étude de ces
opiacés endogènes commence aussi à révéler
les mécanismes par lesquels des facteurs
psychologiques parviennent à moduler notre perception de la douleur,
par exemple dans l'effet
placebo.
Chimiquement, nos opiacés endogènes sont des
petites
protéines (ou peptides), c'est-à-dire des petites chaînes
d'acides aminés qui sont synthétisés au sein même des
cellules nerveuses grâce
à l'ARN messager et aux ribosomes comme toutes les protéines.
Ils sont ensuite transportés le long de l'axone vers les terminaisons nerveuses
où ils sont libérés.
Pour désigner les peptides
endogènes ainsi produits, on utilise le terme général "
d'endorphine " qui fait référence à leurs effets similaires
à la morphine. On en connaît plus d'une vingtaine de représentants
dans le cerveau humain. La liste non exhaustive ci-bas présente ces principales
endorphines ainsi que les protéines dont elles sont extraites. En effet,
toutes les endorphines sont produites en coupant de plus longues protéines
qu'on appelle " précurseurs ".
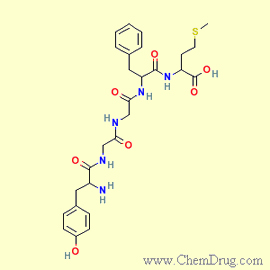
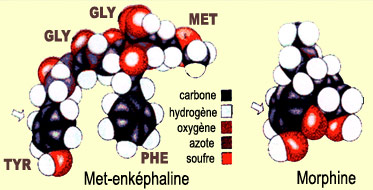
Les
enképhalines, plus précisément la met-enképhaline
et la leu-enképhaline, sont
les deux premières endorphines à avoir été identifiées.
Les deux peptides sont formés d'une chaîne de 5 acides amines, dont
les 4 premiers sont identiques. Seul diffère le dernier qui est de la méthionine
dans le cas de la met-enképhaline et de la leucine dans celui de la leu-enképhaline.
Les
enképhalines sont obtenues par le clivage d'une protéine précurseure
appelée proenképhaline. Chaque proenképhaline contient au
moins sept peptides actifs, dont quatre met-enképhalines et une leu-enképhaline,
qui sont libérés après que des enzymes dites de maturation
aient fait leur travail de coupe.
En
comparant la forme des opiacés endogènes à celle de la morphine,
on note une région similaire dans les deux molécules qui explique
leur affinité commune pour nos récepteurs opioïdes.
Les
enképhalines sont sécrétées dans toutes les structures
centrales et périphériques à proximité des récepteurs
opioïdes mu et/ou delta qui sont leur récepteur naturel. Peu de
temps après leur action, ces modulateurs naturels de la douleur sont inactivés
par clivage par des enzymes de la famille des métallopeptidases.
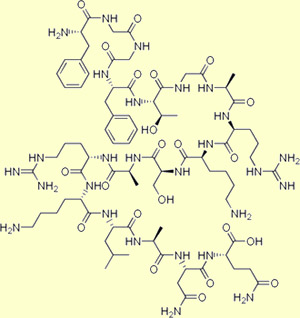
Les
dynorphines, dont l'étymologie grecque renvoie à leur puissance
(dynamis, en grec), est une classe d'opioïdes endogène qui joue un
rôle important dans la modulation de la douleur.
Elles
dérivent de la protéine précurseur prodynorphine qui, lorsqu'elle
est coupée par la proprotéine convertase 2 (ou PC2), libère
plusieurs peptides opioïdes actifs dont la dynorphine A, la dynorphine B,
l'alpha et la bêta néoendorphine. Ces 4 peptides possèdent
la suite exacte des acides aminés de la leu-enképhaline, mais avec
en plus 12, 8, 5 et 4 autres acides aminés respectivement.
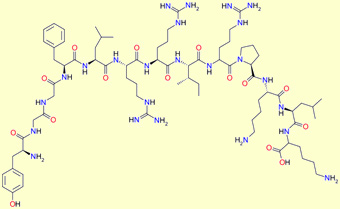
Dynorphin A (1-13) : Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Ile-Arg-Pro-Lys-Leu-Lys
Dynorphin B : Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Gln-Phe-Lys-Val-Val-Thr
a-Neoendorphin : Tyr-Gly-Gly-Phe-Leu-Arg-Lys-Tyr-Pro-Lys
b-Neoendorphin : Tyr-Gly-Gly-Phe-Leu-Arg-Lys-Tyr-Pro
Bien que les dynorphines soient largement distribuées
dans le système nerveux central, on en retrouve de
grandes concentrations dans l'hypothalamus, le tronc cérébral
et la moelle épinière. Elles ont différentes
actions physiologiques selon leur site de production et se
fixent surtout sur les récepteurs
opioïdes kappa (bien qu'elles aient également
une bonne affinité pour les récepteurs mu et
delta).
Le terme générique d'endorphine désigne
aussi certains peptides opioïdes particuliers. On les
distingue cependant par l'ajout d'une lettre grecque devant
le mot. Ainsi, la plus importante se nomme bêta-endorphine.
En plus de diminuer substantiellement la douleur (son pouvoir
analgésique est plusieurs fois supérieur à
la morphine), la bêta-endorphine est le peptide opioïde
qui produit la plus grande sensation d'euphorie. Il est donc
largement produit durant l'exercice
physique soutenu et produit cette sensation en se fixant
sur des récepteurs
opioÏdes de type mu.
Bêta-endorphine
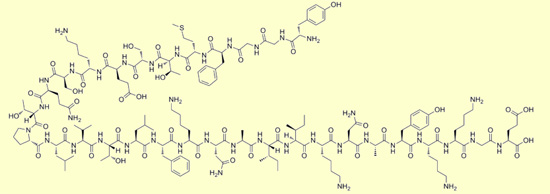
Bêta-endorphine : Tyr Gly
Gly Phe Met Thr Ser Glu Lys Ser Gln Thr Pro Leu Val
Thr Leu Phe Lys Asn Ala Ile Ile Lys Asn Ala Tyr Lys
Lys Gly Glu
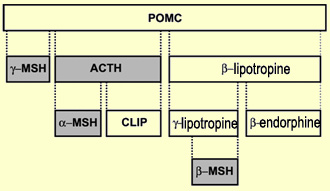
Le précurseur de la bêta-endorphine,
la pro-opiomélanocortine (ou POMC), a ceci de particulier que son clivage
ne produit pas seulement de la bêta-endorphine et d'autres peptides opioïdes,
mais aussi plusieurs autres hormones peptidiques. Selon le type de tissu où
le gène de la POMC va s'exprimer, il sera coupé différemment
et produira des peptides particuliers.
Ainsi,
dans l'hypophyse antérieure, il donnera de la bêta-endorphine, de
la bêta-lipotropine (lié au métabolisme des lipides) mais
aussi l'hormone
adrénocorticotrope (ou ACTH) sécrétée en réponse
à un stress. Mais lorsque le gène de la POMC est traduit dans
les mélanocytes de la peau, elle produira plutôt l'hormone mélanotropine
qui déclenche la synthèse du pigment mélanine qui colore
la peau en réponse aux rayons solaires.
Résumé
des dérivés possibles de la POMC.
La POMC,
que l'on retrouve aussi dans l'hypothalamus, est une chaîne de 241 acides
aminés qui peut être scindé à différents endroits
par de enzymes appelées prohormone convertases. L'un de ses dérivés
de 90 acides aminés, la bêta-lipotropine, avait déjà
été isolée en 1964 par le biochimiste C. H. Li. Mais ne sachant
quelle fonction lui attribuer, celle-ci était demeurée sur les tablettes
pendant 7 ans avant qu'il ne comprenne qu'elle était, entre autres, un
précurseur de la bêta-endorphine. Celle-ci, avec ses 31 acides aminés,
est la plus longue de sa famille qui compte aussi l'alpha, la gamma et la sigma-endorphine.
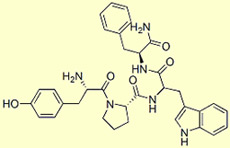
Endomorphine
1
Endomorphine 1 : Tyr-Pro-Trp-Phe-NH2
Endomorphine
2 : Tyr-Pro-Phe-Phe-NH2
Les neurones
de l'hypothalamus contiennent également une autre classe de peptides opioïde
découverte à la fin des années 1990. Il s'agit de l'endomorphine
1 et 2, deux petits peptides de 4 acides aminés chacun qui ont la plus
grande affinité connue avec le récepteur
opioïde de type mu.
Le nom d'endomorphine
(et non d'endorphine, comme dans la bêta-endorphine) rappelle que ces protéines
sont nos ligands naturels qui se fixent sur les mêmes récepteurs
qui permettent à des substances exogènes comme la morphine de produire
leurs effets.
La distribution anatomique des
endorphines suggère un rôle dans le contrôle de la douleur,
la réponse au stress,
l'éveil
et la récompense.
C'est
à peu près à la même époque que l'orphanine
FQ (ou nociceptine) fut découverte. Ce peptide de 17 acides
aminés, dérivé du précurseur prépronociceptine,
n'agit que très peu sur les récepteurs opioïdes classiques.
Il se fixe plutôt sur une classe de récepteurs opioïdes atypique,
les récepteurs
"orphelins" (d'où son nom).
Le
mode d'action de l'orphaline dans la perception de la douleur est complexe puisque
son action peut être parfois analgésique et parfois anti-analgésique
(en bloquant l'action d'autres peptides opioïdes).
Enfin,
parmi tous les autres peptides opioïdes connus, mentionnons la nocistatine
qui dérive elle aussi du précurseur prepronociceptine et serait
impliquée dans la transmission de la douleur, la
mémoire et l'apprentissage.