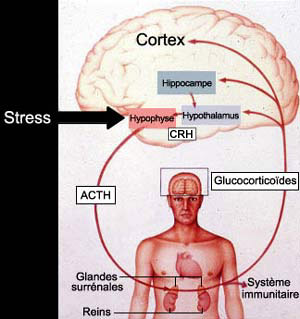
De la perception du danger à la sécrétion d’hormones
préparant l’organisme à y faire face, la réponse met
successivement en jeu : 1) le système limbique, 2) l’hypothalamus,
3) l’hypophyse et 4) les glandes surrénales. Celles-ci sécrètent
des glucocorticoïdes (comme le cortisol chez l’humain, par exemple)
qui vont interagir avec les récepteurs à la sérotonine du
cerveau.
Quand quelqu’un subit un événement
stressant, son taux de glucocorticoïdes sanguin augmente.
Le stress entraîne une activation de l’hypothalamus
qui sécrète alors l’hormone CRH (pour
«corticotropin-releasing hormone»). Cette hormone
amène à son tour l’hypophyse à
produire l’hormone ACTH (adrénocorticotropine)
qui circule dans le système sanguin et atteint les
glandes surrénales où elle provoque le relâchement
de cortisol.
Ce processus forme une
boucle de rétroaction négative où l’excès de
cortisol active les récepteurs aux glucocorticoïdes du cerveau et
supprime la production de CRH. Chez les patients déprimés cependant,
cette boucle ne fonctionne plus d’où une production excessive de
CRH, et donc de cortisol. Plusieurs patients sérieusement déprimés ont un taux
de cortisol sanguin élevé provoqué par un stress chronique.
Le stress chronique et/ou un haut taux de
glucocorticoïdes chez le rat altère certains de ses
récepteurs sérotoninergiques (augmentation des récepteurs
corticaux 5-HT2a et diminution des récepteurs 5-HT1a dans
l’hippocampe). Ces mêmes changements sont observés
chez des humains victimes de suicides
ou de maladies provoquant une hypersécrétion de glucocorticoïdes.
Or, l’administration chronique d’antidépresseurs
provoque les changements opposés sur les récepteurs
sérotoninergiques à ceux produits par un stress chronique.
Elle renverse aussi l’hypersécrétion des hormones
du stress.
Par ailleurs, plusieurs récepteurs aux glucocorticoïdes
(RG) ainsi qu’aux minéralocorticoïdes (RM) (voir
encadré) sont situés dans l’hypothalamus et
l’hippocampe, deux structures impliquées dans le contrôle
de notre humeur et notre capacité à ressentir du plaisir.
Ces récepteurs étant sensibles à la fois au
niveau et à la durée d’activation des différents
corticostéroïdes dans l’organisme, leur dynamique
d’activation aura des répercussions directes sur la
réponse comportementale adoptée.
Par exemple, les corticostéroïdes circulant à
un bas taux facilitent, via les récepteurs MR, les
réactions d’orientation et de paralysie momentanée
associée à la peur. À de hauts taux de
circulation, comme lors d’un stress chronique, c’est
plutôt l’apprentissage de l’inefficacité
de l’action qui est potentialisée via les récepteurs
GR.
Un stress chronique prolongé semble d’ailleurs altérer
la réponse des récepteur MR et GR et a des conséquences
très néfastes sur l’équilibre mental
d’une personne, plus particulièrement lorsque le support
social ou familial est absent. Dans ces conditions, la réponse
à l’origine très adaptative des glucocorticoïdes
devient carrément mésadaptée.
On sait depuis longtemps que les
personnes dépressives montrent une hyperactivité de l’axe
hypothalamo-hypophysio-surrénalien (ou HPA) (voir le texte à droite
de l'image). Par ailleurs, on sait aussi qu’un état d’inhibition
de l’action qui se prolonge favorise l’émergence d’un
état dépressif. Ce stress chronique, en sollicitant exagérément
l’axe du stress (HPA), amèneraient des changements structuraux dans
certaines régions du cerveau. C’est le cas par exemple de l’hippocampe
dont la région CA3 subit des pertes neuronales importantes sous l’effet
d’un stress prolongé.
D’autres études ont
aussi rapporté une diminution du nombre de récepteurs aux glucocorticoïdes
dans l’hippocampe et le cortex préfrontal chez des victimes du suicide.
Bien qu’il soit difficile de savoir si ces changements structuraux sont
d’origine génétique où le résultat d’un
activation chronique de l’axe HPA, il demeurent cohérent avec l’hyperactivité
de cet axe dont le frein naturel se trouve ainsi diminué.
Autre
exemple : chez les gens atteints du syndrome de Cushing, une maladie où
un excès de cortisol est produit, on retrouve une incidence élevée
de dépression. De plus, leur dépression cesse lorsque leur taux
de cortisol se normalise avec les traitements.
Tout porte donc à
croire que le produit final de l’axe HPA, les glucocorticoïdes, jouent
un rôle dans l’état dépressif en influençant
plusieurs systèmes de neurotransmetteurs dont la sérotonine, la
noradrénaline et la dopamine, tous trois impliqués dans la dépression.
On donne le nom de corticostéroïdes
(ou corticoïdes) aux hormones sécrétées
par la région externe des glandes surrénales.
Ils peuvent être séparés en trois groupes
qui ont chacun leurs récepteurs distincts : les androgènes,
impliquées dans le développement des caractères
sexuels; les minéralocorticoïdes (aldostérone,
corticostérone, désoxycortisone) qui régulent
l'équilibre osmotique du corps; et les glucocorticoïdes
(cortisone, hydrocortisone, prednisone) qui, outre leur
activité anti-inflammatoire et immunosuppressive, stimulent
la synthèse du glucose et augmentent la mobilisation
des acides gras et des protéines pour répondre
à la demande métabolique plus élevée
engendrée par un stress.
Les glucocorticoïdes jouent un rôle extrêmement
important dans les réactions de peur, d’anxiété
et dans les états dépressifs. Ils exercent souvent
leurs effets sur le comportement en augmentant ou diminuant
l’efficacité de certaines voies neuronales.
On distingue souvent deux phases
dans un traitement aux antidépresseurs. Durant les 15 premiers jours, l’état
dépressif du patient ne s’améliore pas vraiment. À
partir de deux à trois semaines cependant, le patient retrouve progressivement
le sommeil, l'appétit, un regain d'énergie et des pensées
positives. On recommande alors de poursuivre le traitement pendant plusieurs mois
pour minimiser le risque des rechutes.
Différentes hypothèses
ont été formulées pour expliquer ce délai. On pense
qu’au début du traitement, suite à l'inhibition de la recapture
de la 5-HT, les autorécepteurs sont rapidement saturés de sorte
que c’est l’effet inhibiteur des autorécepteurs qui prédomine
ce qui diminue la libération de sérotonine dans la fente synaptique.
Puis, dans un deuxième temps, les autorécepteurs finissent par être
désensibilisés et les potentiels d’action sont produits plus
facilement par le neurone pré-synaptique. La sérotonine n'étant
pas recaptée à cause des antidépresseurs, sa concentration
extracellulaire augmente, et la transmission sérotoninergique est facilitée.
L'effet des antidépresseurs
peut se rapprocher de celui de l'ecstasy
qui provoque le relâchement de grandes quantités de sérotonine
aux terminaisons nerveuses des neurones. C'est cet excès de sérotonine
que l'on suspecte d'être à l'origine des effets psychiques particuliers
de l'ecstasy, dont ceux reliés au sentiment de bien-être. Un effet
qui va donc dans le même sens que celui des antidépresseurs.
Une première
hypothèse formulée durant les années 1960 ciblait la noradrénaline
comme le neurotransmetteur principal impliqué dans la dépression.
Cette hypothèse dites «des catécholamines» proposait
que la dépression était due à une déficience en noradrénaline
dans certains circuits cérébraux, alors que la manie correspondait
à une surabondance du même neurotransmetteur. Bien qu’encore
reconnue, cette hypothèse n’explique pas tout, et en particulier
pourquoi des fluctuations du taux de noradrénaline n’affecte pas
l’humeur de certaines personnes.
Durant les années 1970,
l’implication d’un autre neurotransmetteur, la sérotonine,
fut postulée dans ce que l’on a appelé l’hypothèse
«permissive» de la dépression. Celle-ci propose que la diminution
de la quantité de sérotonine à certaines synapses puisse
aussi être à l’origine d’une dépression en déclenchant
ou en « permettant » une baisse de noradrénaline. Par conséquent,
même si l’on reconnaissait toujours un rôle important à
la noradrénaline dans la dépression, on pouvait maintenant agir
sur la sérotonine pour tenter de soulager la dépression. Une voie
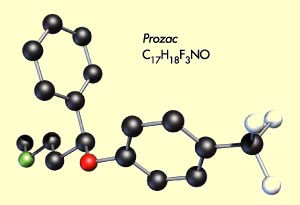
thérapeutique exploitée par le Prozac et tous les autres inhibiteurs
sélectifs de la recapture de la sérotonine (ISRS) depuis les années
1980.
Fluoxétine (Prozac)
Un troisième
neurotransmetteur d'importance dans la dépression est la dopamine.
Cette molécule est aussi impliquée dans la schizophrénie
et la maladie de Parkinson. Elle joue un rôle important dans le renforcement
positif et la récompense, autrement dit dans la poursuite de l'action gratifiante.
L'utilisation de substances dopaminergiques et de stimulants comme antidépresseurs
donne d'ailleurs des résultats positifs et rapides chez plusieurs patients,
ce qui en fait des compléments intéressants aux autres antidépresseurs
qui peuvent prendre plusieurs semaines à agir.
Les médicaments
qui agissent directement sur la dopamine sont cependant plus susceptibles de créer
des dépendances, ce qui rend leur utilisation plus délicate. Plusieurs
drogues comme la cocaïne, les opiacés ou l’alcool augmentent
d’ailleurs la production de dopamine ce qui pourrait expliquer pourquoi
plusieurs dépressifs les consomment.
Un nombre important de chercheurs
croient que l'expression de " déséquilibre chimique "
pour parler des causes physiologiques de la dépression n'est plus vraiment
adéquate.
Cette hypothèse du " déséquilibre
chimique " date des années 1960. Les premiers antidépresseurs
découverts furent les tricycliques et les inhibiteurs de la MAO. Ces molécules,
en plus d'améliorer les symptômes de la dépression chez nombre
de patients, sont reconnues pour augmenter d'une manière ou d'une autre
les taux de dopamine, de noradrénaline et de sérotonine. D'où
l'hypothèse d'un déséquilibre au niveau de ces neurotransmetteurs.
Hypothèse qui fut d'ailleurs plutôt fructueuse au niveau de la recherche
des dernières décennies du XXe siècle. De plus, en attirant
l'attention sur le fait que les troubles de l'humeur pouvaient être reliés
à un dysfonctionnement physiologique et pas seulement à un manque
de volonté ou à une faiblesse du caractère, cette hypothèse
diminuait le sentiment de culpabilité inutile qui habite souvent les personnes
dépressives.
Mais les efforts pour identifier
plus précisément le déséquilibre en question ont donné
des résultats assez décevants et contradictoires. Les recherches
se tournent maintenant davantage vers les récepteurs des neurotransmetteurs
plutôt que sur les neurotransmetteurs eux-mêmes, ainsi que sur les
événements moléculaires qui participent à la régulation
des gènes. Mais encore ici, il y a place à la controverse. En effet,
il y a relativement peu de preuves directes de l'altération des récepteurs
ou d'anomalies de l'expression génique reliée à ces récepteurs
ou d'autres enzymes lors de la dépression. Par ailleurs, le délai
thérapeutique de deux à trois semaines (voir l'encadré à
gauche) entre l'effet des médicaments antidépresseurs sur les neurotransmetteurs
et leurs effets sur l'humeur n'est toujours pas bien compris. En somme, la situation
est beaucoup moins simple qu'on le pensait dans les années 1960 quand on
a formulé l'hypothèse du " déséquilibre chimique
".
Devant ces difficultés à
obtenir des données sans équivoques appuyant cette hypothèse,
certains en sont même venus à se demander si l'usage extensif que
l'on fait toujours de l'expression "déséquilibre chimique"
ne soulevait pas des problèmes éthiques, voire politiques. Aux États-Unis
par exemple, où la publicité pour les antidépresseurs est
permise dans les
grands médias, les compagnies pharmaceutiques n'ont pas toujours
fait dans la nuance. Les publicités simplistes affirmant qu'une substance
de notre cerveau subit un déséquilibre lors de la dépression
et que tel ou tel antidépresseur rétablit comme par magie une situation
d'équilibre idéale n'est sans doute pas étranger au succès
fulgurant qu'ont connus les antidépresseurs de type ISRS (Prozac, Zoloft,
Paxil, etc…). Et aux milliards de profits qu'ils ont générés
pour ces compagnies.
Les antidépresseurs n’ont
pas que des effets au niveau pré-synaptique. Sur les neurones post-synaptiques,
l’action antidépressive des tricycliques et des IMAO peut s’expliquer
par la "down regulation" (diminution du nombre mais non de la sensibilité)
des récepteurs bêta-adrénergiques et des récepteurs
sérotoninergiques 5 HT2 . On observe aussi une désensibilisation
des récepteurs noradrénergiques couplés à l’adénylate
cyclase. Les phénomènes de transduction via les protéines
G couplées aux récepteurs représentent un autre site d’action
post-synaptique possible, comme c’est probablement le cas pour le
lithium.